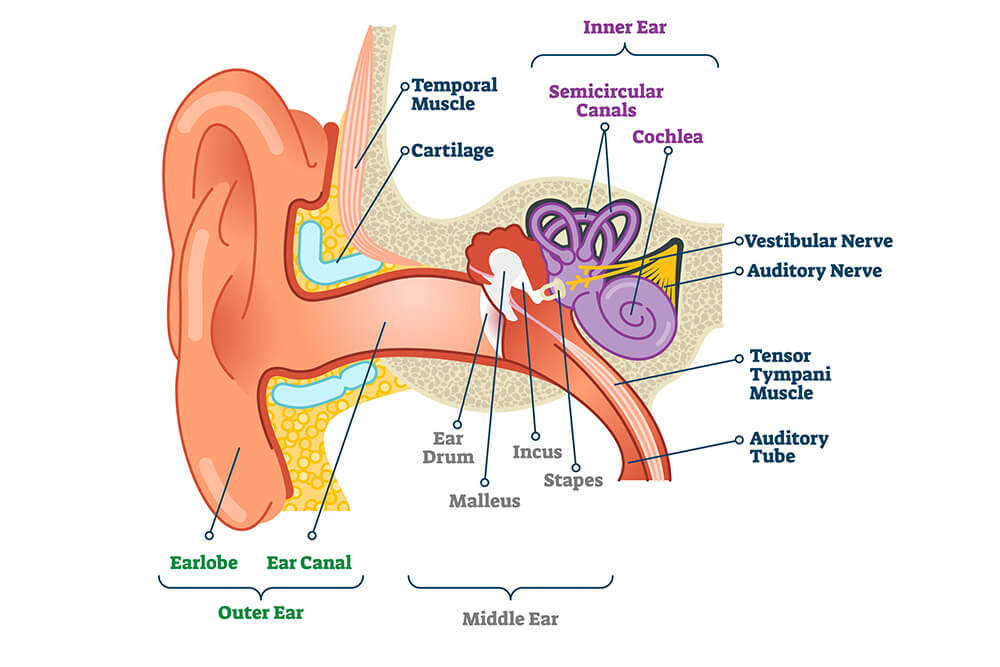
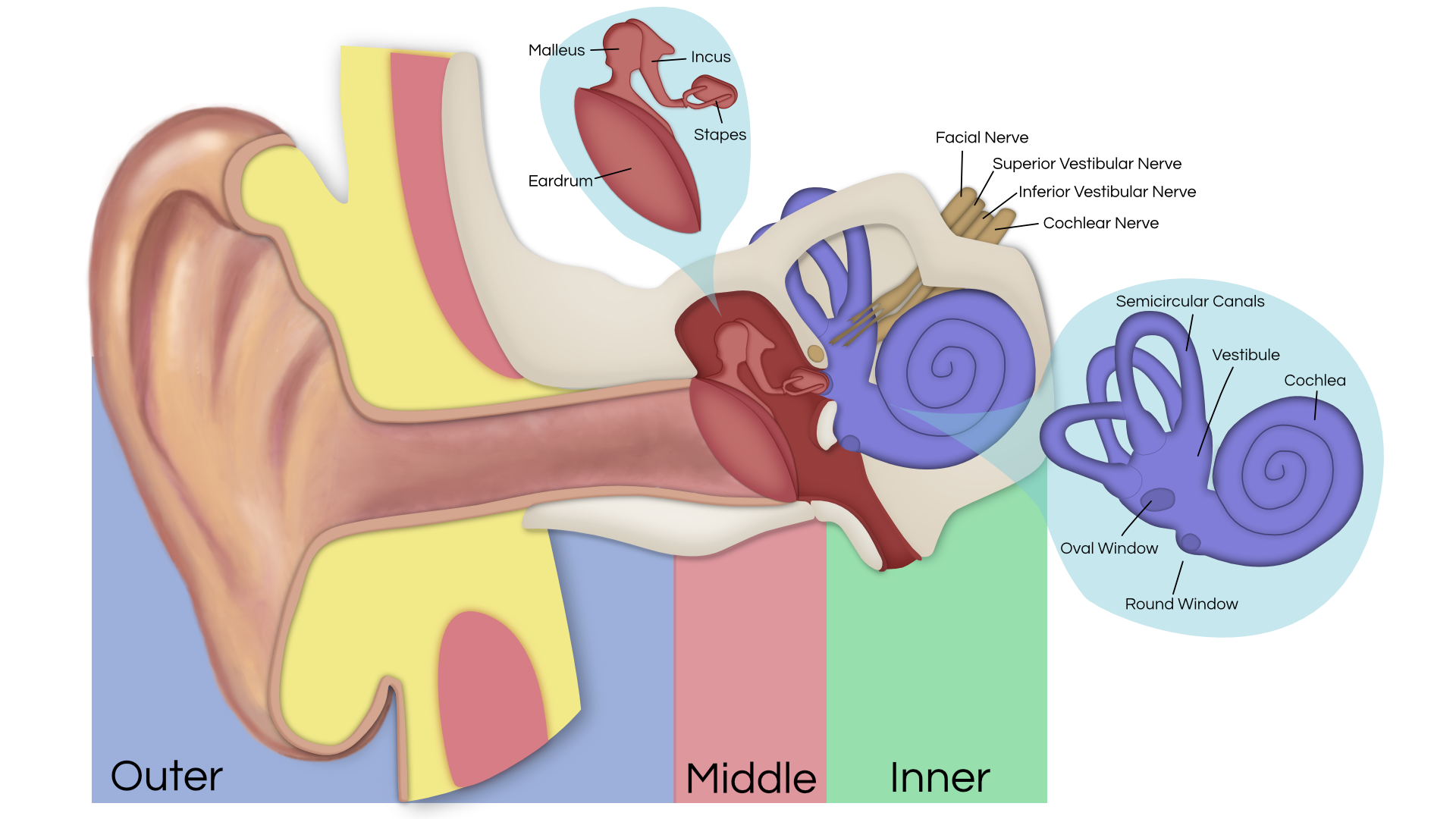
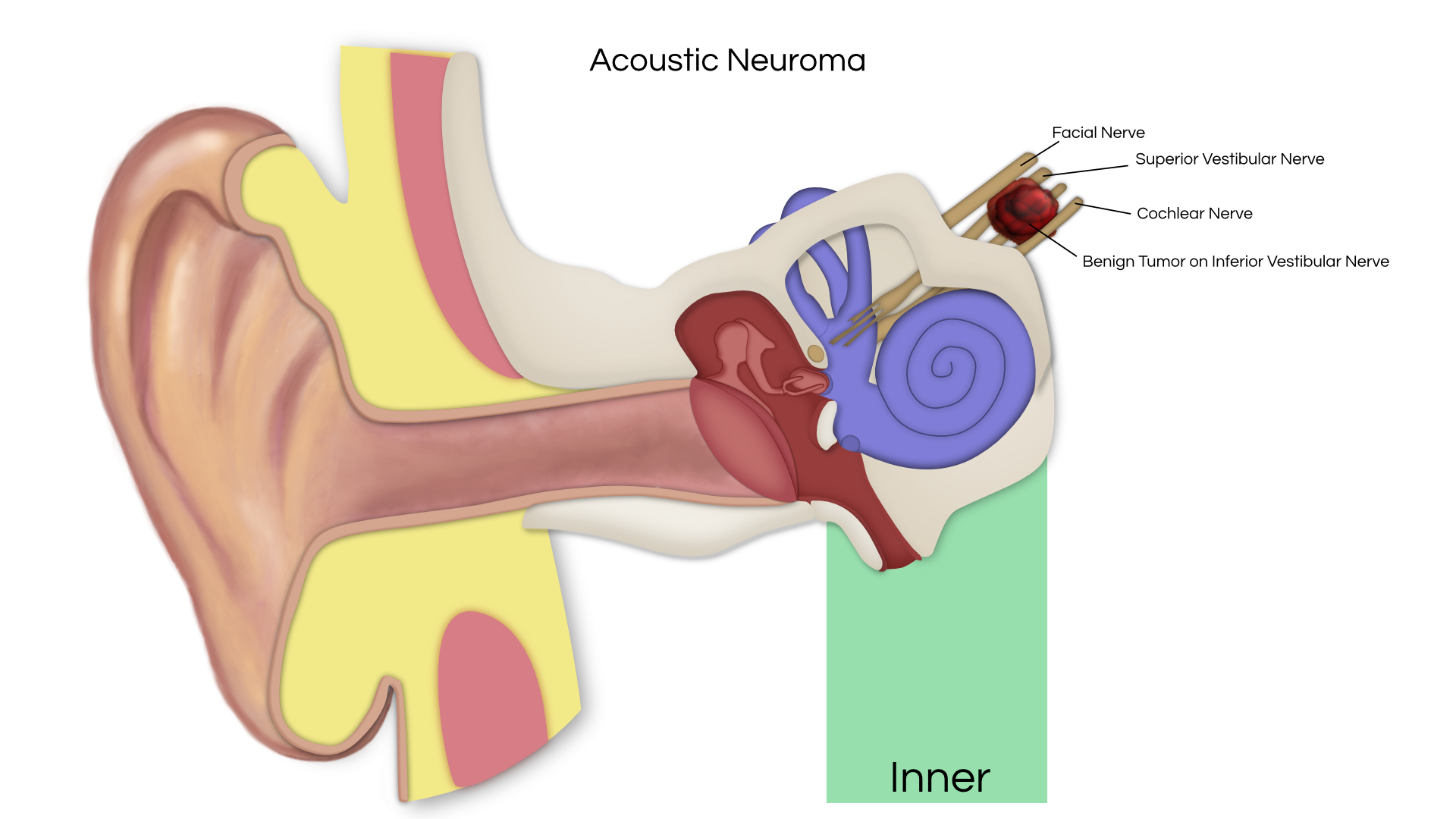
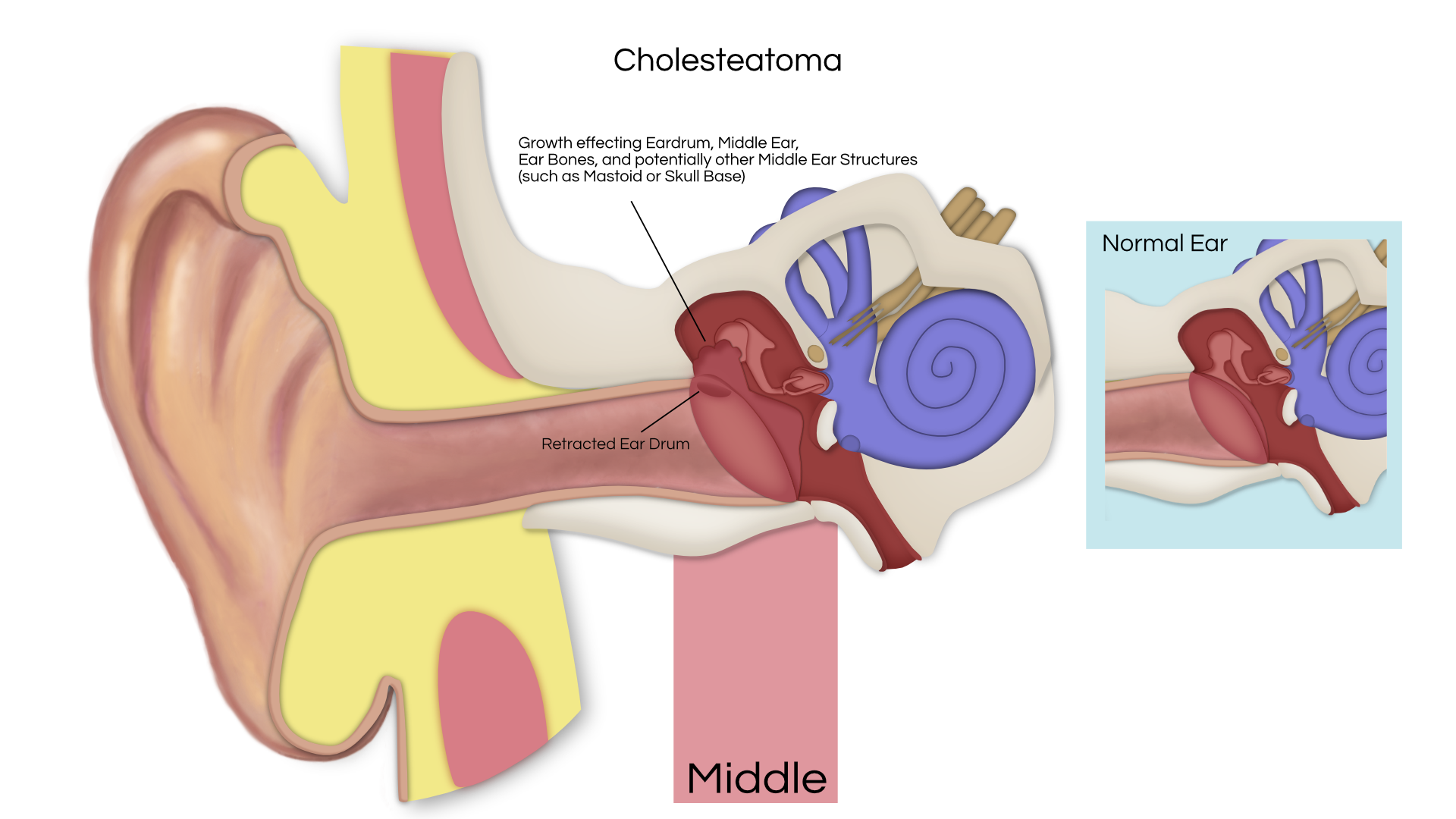
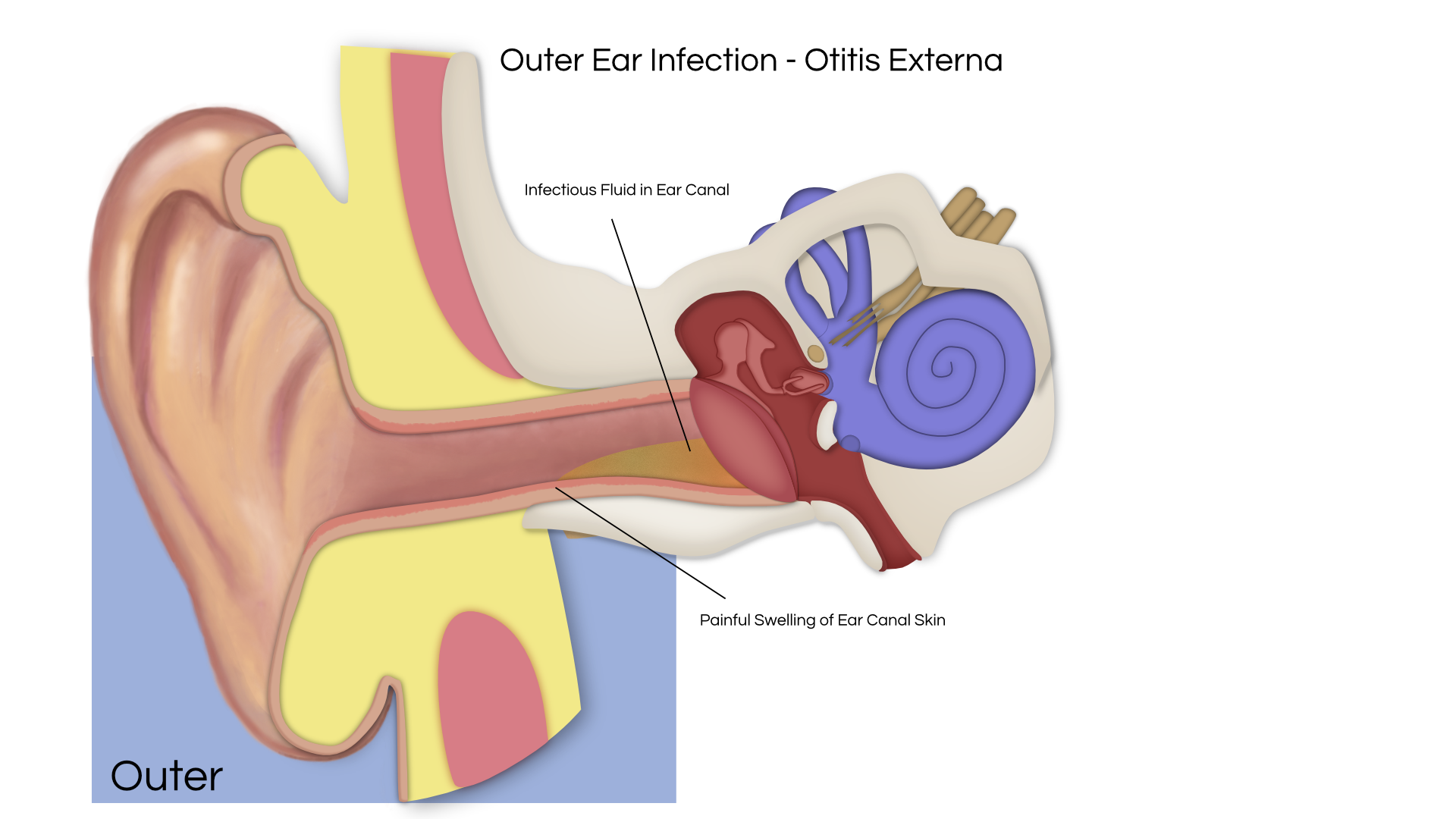
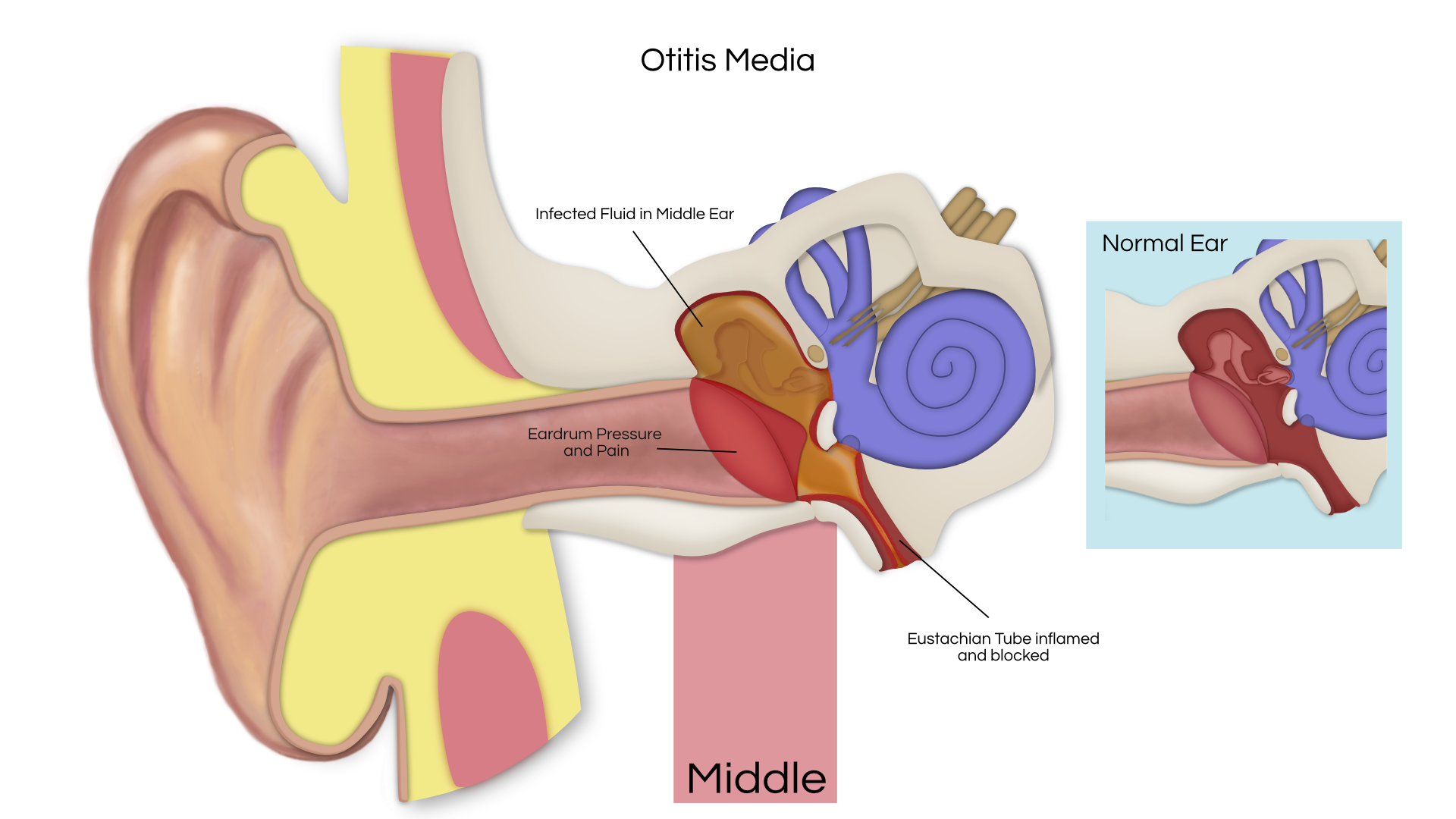
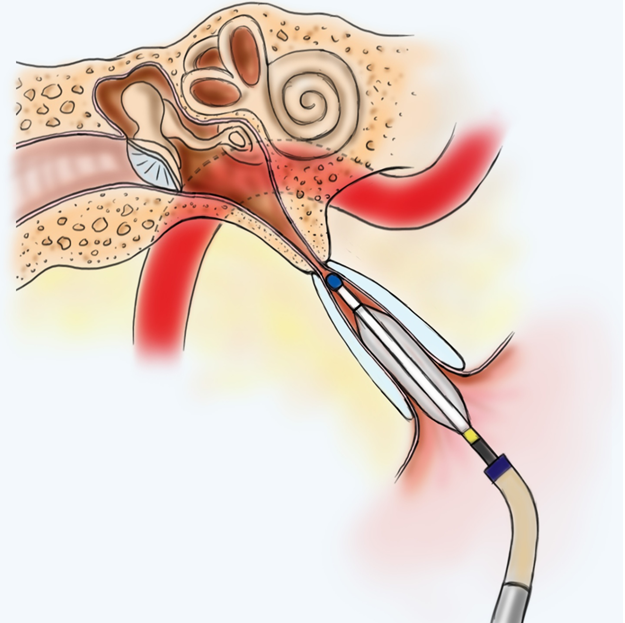
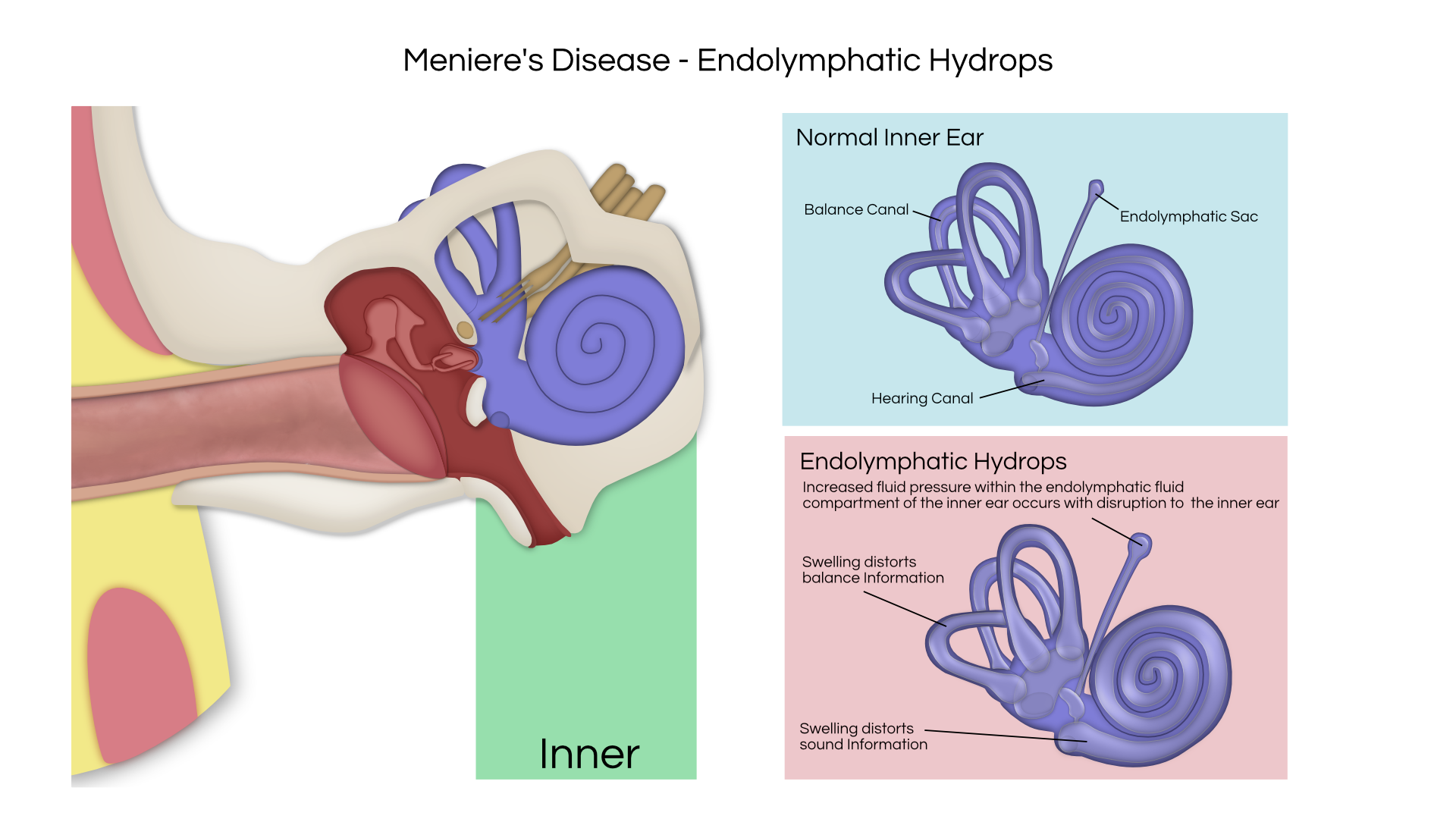
An acoustic tumor (sometimes termed a neuroma or schwannoma) is a benign (non-cancerous) tissue growth that arises on the eighth cranial nerve. Properly, it is referred to as a vestibular schwannoma. These tumors generally cause one or more of the following symptoms: hearing loss, ringing in the ear, and dizziness or unsteadiness. Once diagnosed, three treatment options exist: observation, radiation therapy, and surgery.
An acoustic tumor (sometimes termed a neuroma or schwannoma) is a benign (non-cancerous) tissue growth that arises on the eighth cranial nerve. Properly, it is referred to as a vestibular schwannoma. These tumors generally cause one or more of the following symptoms: hearing loss, ringing in the ear, and dizziness or unsteadiness. Once diagnosed, three treatment options exist: observation, radiation therapy, and surgery.
Surgical Resection
At this time, the only treatment that can cure a patient with an acoustic neuroma is surgical removal. If surgery is selected as the best course of action by the patient and doctors, the main goal is the preservation of life, with a minimum of future physical disturbances. To accomplish this, a team consisting of a Neurotologist, Neurosurgeon, and sometimes Internist, Anesthesiologist, and/or specially trained surgical nurses, provide the surgery as well as the pre-and post-operative care. The Neurotologist and the Neurosurgeon are co-surgeons during this surgical procedure.
Following surgery, the patient is usually admitted to the intensive care unit for 24-48 hours post-operatively for close observation. After that period, the patient is transferred to the regular surgical ward or to an intermediate unit. Patients are usually in the hospital for 4-7 days. Within the last several decades, microsurgical techniques have been pioneered and refined. With the use of an operating microscope and specialized instruments, the Neurotologist and Neurosurgeon are able to remove the tumor with an extremely low mortality rate. Damage to the surrounding nerve tissue is markedly decreased. Routinely, facial nerve function is monitored during surgery. In some situations the cochlear nerve is also monitored when it appears feasible to preserve hearing. The size of tumor, hearing status, patient’s age, and overall health determine which approach is utilized for removing the tumor when surgery is contemplated. When the tumor is confined within the internal auditory canal, or just outside the canal, and there is useful hearing, the middle fossa or retrosigmoid approaches are generally preferred. Both approaches allow the possibility of hearing preservation. When the hearing is poor, the translabyrinthine approach is often selected. The translabyrinthine approach is very effective for tumor removal and preservation of the facial nerve; however, there is no chance for hearing preservation. For larger tumors, occasionally the retrosigmoid approach may be utilized, and sometimes a combined approach (such as middle fossa/retrosigmoid approach) has been utilized when one is trying to preserve hearing.
The treatment of vestibular schwannomas must be individualized to each patient by an experienced team of doctors. Microsurgical removal of these tumors remains the treatment of choice as determined by the National Institute of Health (NIH) Consensus Development Conference. Further research is needed on the advantages and risks of other management options, which include careful observation and stereotactic radiation.
Observation
The natural history of acoustic neuromas is not known. Each tumor has its own biology and growth characteristics. In general, acoustic neuromas are regarded to be slow growing, averaging 2mm of growth per year (range 1mm – 12mm/year). The critical question to be answered when observation is considered is whether the acoustic neuroma will cause any problems during the natural course of the patient’s remaining life span. With increasing patient age and smaller tumor sizes, observation is often selected, especially if slow tumor growth can be documented with serial imaging.
Stereotactic Radiation Therapy
Another form of treatment which has become an alternative to surgery is the use of stereotactic radiation therapy (i.e., gamma knife). Stereotactic radiation therapy is a technique based upon the principle that a single high dose of radiation delivered precisely to a small area of tumor could arrest the growth of the tumor and not damage surrounding brain tissue and/or function. In this technique, under a light general anesthesia, the head is secured in a special device to hold it in a proper position, and a single dose of radiation is applied (stereotactically) by the therapist. The dose is calculated to injure/kill tumor cells yet minimize side effects or risks. There is no incision. Almost all patients are discharged from the hospital the same day or the following day. Life-long follow up with MRI scans is currently recommended. Unlike microsurgery, which would remove the tumor, stereotactic radiation therapy “controls” the tumor growth. The tumor does not disappear but persists, allegedly, in a “harmless” state. The risk of microsurgical removal are well defined, short-term and consistently quantified in thousands of patients over 30 years. No such data exists for acoustic neuroma treatment by stereotactic radiation therapy. Short-term risks of stereotactic radiation therapy appear comparable to surgery in many areas. The real risk of stereotactic radiation therapy may be ongoing, long-term and, as yet, undetermined. Tumor regrowth has been seen in 6 – 24% of stereotactic radiation therapy patients. The long-term growth control rates for stereotactic radiation therapy has not been established. Serious complications can also occur as a result of stereotactic radiation therapy.
Cost for microsurgical removal includes the operation, approximately a seven day hospital stay, and postoperative MRI scanning at 1 and/or 3-5 years. The patient is also cured (disease free) with surgery, unless subtatotal resection is performed. Cost for stereotactic radiation therapy includes the treatment and MRI scans, which are required at 6, 12, 24, 48, 96, etc. months, since the tumor persists. There are medical costs associated with this follow up. When the tumor regrows, the substantial cost of salvage surgery must be factored. There is also evidence that suggests salvage surgery in a patient who has had prior stereotactic radiation therapy is much more difficult with decreased chance of facial nerve preservation. Radiation therapy represents an alternative for patients who are elderly, or have medical problems such as cardiac or pulmonary disease and are at high risk for surgical removal.
Size of Tumor
Risks and complications of surgery vary with the size of the tumor. Larger tumors have more serious potential complications, and more likelihood of complications. In addition, the approach to remove the tumor may differ, depending upon the size of the tumor and the patient ’s medical status, age, and hearing status. The removal of an acoustic tumor is a major surgical procedure, entailing a craniotomy (opening into the skull), with possibilities of serious complications, even death, whether the tumor is large or small. Different approaches are used (as indicated above) to minimize the complications and, at the same time, remove the entire tumor. Tumors are classified as small, medium, or large.
Small Tumor
The small tumor is one in which the tumor is confined within the bony canal extending from the inner ear toward the brain. Contained in this canal are the hearing and balance nerves, as well as the facial nerve, which enables the face to move on that side. In addition, there are numerous blood vessels which supply the inner ear.
The operation for acoustic tumors is performed under general anesthesia, using an operating microscopic. The surgical approach to a small tumor may be by the middle fossa approach, through an incision in front of and above the ear; by the suboccipital approach, with an incision toward the back of the head; or by a translabyrinthine approach, with the incision behind the ear. The approach is determined by the amount and quality of hearing in each patient.
In the vast majority of cases of small tumors, the tumor is totally removed. When deemed feasible, every effort is made to preserve the hearing in small tumors by one of the first two approaches, and still remove the tumor. However, it is imperative that tumor removal take precedence over hearing preservation. In approximately 35% of the cases, serviceable hearing may be preserved. If the tumor involves the hearing nerve or an artery leading to the inner ear, total loss of hearing will result in the operated ear.
Medium Tumor
In a medium tumor, there is extension from the bony canal towards the brain, but there is not yet pressure on the brain itself.
Again, the surgery is performed under general anesthesia, utilizing an operating microscope and surgical team. Frequently a translabyrinthine or suboccipital approach is utilized. If a translabyrinthine approach is selected, the incision is made behind the ear, over the mastoid bone. The mastoid and inner ear structures are removed to expose the tumor. The tumor is then totally removed in the vast majority of cases. The mastoid bone defect is closed with fat taken from the abdomen. This approach sacrifices the hearing and balance nerves in the operated ear, and the patient is made permanently deaf on that side. This approach has been developed to provide maximum safety to the facial nerve, and allow complete removal of the tumor. The balance mechanism of the opposite ear usually provides stability for the patient in approximately one to four months.
Large Tumor
The large acoustic tumor has extended out of the bony canal, and into the brain cavity, producing pressure on the brain and vital structures. The approach to a large acoustic tumor requires more extensive removal of the bone, to expose the tumor and control large vessels, which obstruct access to the tumor. Occasionally, special vascular studies are required prior to surgery to help diagnose the location of larger vessels.
The surgical approach, the translabyrinthine and/or suboccipital approach, is done through an incision behind the ear. The mastoid bone, inner ear structures, and a portion of the skull are removed to expose the tumor. The tumor is then totally removed, unless vital sign changes prevent its removal from the brainstem. If there are changes in blood pressure, pulse rate, or respiratory rate, the surgery may have to be terminated before the tumor is totally removed. In this instance, it would be necessary to have a second operation to completely remove the tumor. The surgical defect is closed with fat taken from the abdomen. The translabyrinthine-suboccipital approach causes the patient to be permanently deaf in the operated ear. The balance mechanism is removed, but the balance mechanism of the opposite ear will provide stabilization of the patient in one to four months.
Full Versus Partial Removal
The goal of acoustic tumor surgery is to remove the entire tumor and preserve as much function as possible. Partial tumor removal may be necessary if the patient’s response to surgery indicates disturbances of any vital brain centers, such as respiration, blood pressure, or heart function. Most of the time, if there is a disturbance in the brain center and surgery is stopped, function can be restored. Occasionally, however, once the vital brain centers are disturbed, they do not recover.
If termination of the operation is necessary without removal of the entire tumor, the remaining portion of the tumor may gradually enlarge to again produce symptoms. A subsequent operation can often then be accomplished without a significant change in vital signs, as there is a change in the relationship of the tumor to the brain.
If the tumor is known to be partially removed, you will be so informed. As mentioned above, reduction of the size of the tumor often allows it to separate from the vital brain centers, and it can be removed at a later date. In most cases, a wait of two weeks to four months is indicated, depending upon the circumstances. In other cases, a course of continued observation is determined best for the patients. If this is necessary, x-ray or MRI imaging techniques will be used to evaluate the tumor from time to time for possible regrowth.
Risk and Potential Complications
Summary
There is no known “cure” for an acoustic neuroma at the present time except surgical removal. The earlier tumors are diagnosed and removed, the less likely the possibility of serious complications.
It is important to face the problem of an acoustic neuroma as it exists at the time of diagnosis, and accept whatever risks are associated with the chosen course of treatment. Maintaining a positive attitude before, during, and after surgery is necessary for the patient, as well as all members of the surgical team, for the best results.
This summary is based on our personal experience in managing a large series of acoustic tumor patients. If you have any questions pertaining to the problem of your tumor, please feel free to discuss them with our office. If you wish further consultation with another surgeon regarding this situation, feel free to do so. We will be pleased to send copies of our office records pertaining to your case to any consultant you may desire.